Latest News
- Clues beginning to emerge on asymtomatic SARS-CoV-2 infection
- Back in November of 2020, during the first wave of the COVID-19 pandemic, I was teaching an in-person microbiology laboratory. One of my students had just been home to see his parents, and they all c…
- Read more
- Could there maybe be better uses of genetics and probiotics?
- Professor Meng Dong and his laboratory have created a probiotic that can metabolize alcohol quickly and maybe prevent some of the adverse effects of alcohol consumption. The scientists cloned a highl…
- Read more
- ChatGPT is not the end of essays in education
- The takeover of AI is upon us! AI can now take all our jobs, is the click-bait premise you hear from the news. While I cannot predict the future, I am dubious that AI will play such a dubious role in…
- Read more
- Fighting infections with infections
- Multi-drug-resistant bacterial infections are becoming more of an issue, with 1.2 million people dying of previously treatable bacterial infections. Scientists are frantically searching for new metho…
- Read more
- A tale of two colleges
- COVID-19 at the University of Wisconsin this fall has been pretty much a non-issue. While we are wearing masks, full in-person teaching is happening on campus. Bars, restaurants, and all other busine…
- Read more
16-5 Putting It All Together - Examples of Responses of the Immune System to Different Types of Pathogens
( 36666 Reads)
|Learning Objectives
- Be able to describe how the entire immune system responds to the attack of a bacterial pathogen.
- Be able to describe how the entire immune system responds to the attack of an intracellular parasite.
Chapter 15 and the previous sections in this chapter have introduced the various parts of the immune system and their reactions, but it is helpful to rethink their functions in the context of a complete response to an invading pathogen. In this section, we will examine how the immune system as a whole recognizes and repels an invader and in the process give you a better look at the forest of host defense and not just the trees.
Microbial Pathogens
In this example, we will examine the response of the immune system to Streptococcus pyogenes, a common pathogen of humans. S. pyogenes is responsible for strep throat, scarlet fever, impetigo and (rarely) severe invasive infections, the latter earning it the name flesh-eating bacteria from the media. We will assume that the host has not been previously exposed to this strain of S. pyogenes. Here we will examine one hypothetical scenario, but it is by no means the only outcome of infection with this organism. The process of infection with S. pyogenes and the body's immune response is summarized in Figure 16.12.
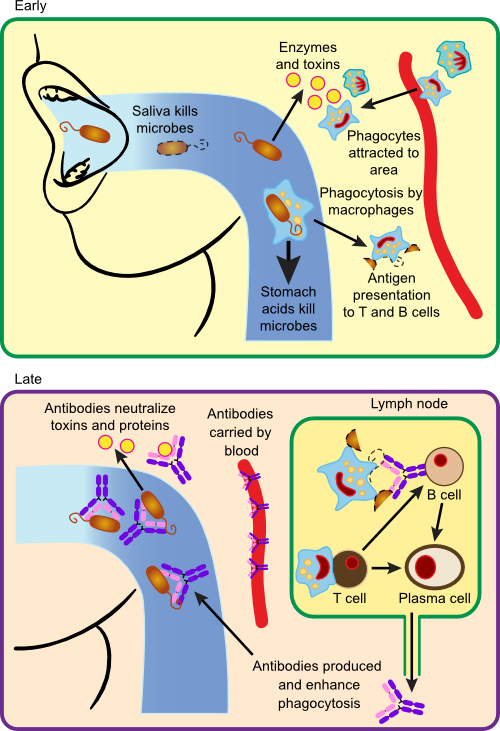
Figure 16.12. Immune Response to Bacterial Infection with S. pyogenes.. Examples of the range of immune defenses against a bacterial pathogen include antibodies and phagocytes that capture and kill the invading microbe. Most of the destruction occurs in the extracellular fluid.
Infection by S. pyogenes typically begins in the mouth and upper respiratory tract. In this example imagine that a group of 10,000 or so S. pyogenes organisms enter the mouth from a droplet of mucus picked up from a nearby child ill with strep throat. Many of them are destroyed by the stomach juices when swallowed and still others are digested by lysozyme in saliva that degrades their outer peptidoglycan layer. The microbes that are left attempt to attach to the surface of the throat using lipoteichoic acid present in their cell walls, but the normal flora occupy most sites for attachment. Many are also repelled by secretory IgA antibody in saliva that cross-reacts with antigens on their surface. However, a few are successful in gaining a loose attachment in the throat and they then bind more tightly with the help of M protein on the microbe and fibronectin present on the epithelial cell surface. Once bound, the microbes begin to multiply and increase in number. Multiplication is hampered by the low pH created by the normal flora and the presence of β-lysins. Lysozyme is also constantly chewing at the cell walls of attached bacteria, but the presence of the capsule protects them from some of these attacks. The colony begins to expand and invade layers below the epithelial cells. The host's surface defenses have failed to repel the infection.
S. pyogenes begins secreting a damaging cocktail of enzymes into the surrounding host tissue. Hyaluronidase degrades hyaluronic acid, a compound that cements cells together in many mammalian systems, allowing further invasion. Secretion of DNase, RNAse and proteases also aid the microbe. This internal invasion causes the secretion of bradykinin and other chemokines from damaged cells, thus initiating the process of inflammation. Bradykinin binds to receptors on blood vessel cells in nearby capillaries causing them to create gaps in the blood vessel walls. Microbial products and the chemokines from damaged cells leak into the surrounding area and into the bloodstream, attracting the attention of phagocytes (mostly neutrophils, but also macrophages) and they begin to migrate to the area and attack the invading S. pyogenes.
At this point phagocytic attack is inefficient, because the bacterium secretes several proteins such as leukocidins, that are toxic to phagocytes. S. pyogenes is also surrounded by a capsule made of hyaluronic acid, yes, the same compound cementing our cell, and this disguise makes the microbes difficult to recognize. Interestingly, this capsule is also attacked by hyaluronidase made by the pathogen, but they seem to make enough of it that it is still protective. The capsule also makes phagocytosis difficult. Despite the resistance of S. pyogenes, a significant number are phagocytized and killed by neutrophils and macrophages. However, due to the slow rate of phagocytosis, the number of bacteria continues to increase. The body attempts to wall off the infection by encasing it in a fibrin clot, but streptokinase produced by the microbe dissolves the clot, further advancing the infection.
As the number of microbes increases, greater numbers of neutrophils penetrate the area. Antigens are also picked up by macrophages and dendritic cells. Active phagocytosis causes the release of more mediators of inflammation and the inflammatory response intensifies. This now becomes noticeable as soreness at the back of the throat. As neutrophils fill with bacteria, they die and make up the pus that runs down the back of the throat and causes the characteristic yellow spots of strep throat.
Bacteria taken up and destroyed by macrophages and dendritic cells are processed and protein fragments, coupled with MHC II molecules are displayed on the cell membrane. During this processing, the macrophages and dendritic cells move toward a nearby lymph node. Inside the lymph node, processed antigens lying in the cleft of the MHC II molecule are presented to a series of T helper cells. Eventually, a successful interaction between the TCR of a TH2 cell and the antigen-presenting cell causes the release of IL-1 and IL-6 from the macrophage. One result of the increased levels of IL-1 is its interaction with the hypothalamus in the brain, which then increases the temperature of the body. The characteristic fever of strep throat increases the activity of T cells. IL-1 and IL-6 stimulate the T cells to proliferate. Proliferating T cells produce IL-2 and IL-4-6 and these further induce T cell multiplication. Most of the TH2 cells help B cells produce antibody, but some remain as memory cells and circulate through the blood and lymph systems.
Receptors on B cells begin to encounter antigens that are flowing from the infected area into the lymph node and these reactions also cause them to proliferate. Some of the antigen that binds to the B cell ends up in MHC II molecules on the surface of the B cell and is presented to T cells residing in the lymph nodes. A previously activated TH2 cell recognizes this antigen in the MHC II molecule and secretes IL-1, IL-4 and IL-5 that stimulate B cells to multiply. As the number of B cells increases, TH2 cells start secreting IL-2, IL-4 and IL-6 that signal to the B cells to differentiate into plasma cells. Plasma cells mobilize large numbers of structures needed for the production and secretion of antibodies (e.g., ribosomes, endoplasmic reticulum, Golgi apparatus etc.). Mature plasma cells migrate to the center of the lymph node and begin secreting IgM antibody specific to the antigen. The rate of secretion can exceed 10 million antibody molecules per minute from an active plasma cell. A fraction of the B cells do not differentiate, but remain as B memory cells. These memory cells disseminate throughout the body in lymphoid tissues and serve as sentries waiting for the next invasion by S. pyogenes.
The full process of antibody induction takes around 7 to 14 days. Initially IgM is made, but this shifts to IgG later in infection. Antibody now exits by the efferent duct, travels through the lymph system and enters into the bloodstream. It circulates in the body and eventually comes in contact with S. pyogenes in the throat.
The entrance of antibody into the sites of infection turns the tide in favor of the host. A large assortment of antibodies is made to many different antigens present on the microorganism. Antibodies directed against the toxins of the microorganism react with and neutralize them. Antibodies against cell surface antigens stick to the microbe and attempt to induce the complement cascade. In the case of S. pyogenes, this reaction is not very effective due to the thick capsule surrounding the microbe. However, antibodies directed against the M protein are extremely effective opsonins. Phagocytes can now rapidly engulf and phagocytize S. pyogenes. Entrance into the phagocyte is rapidly lethal for S. pyogenes, because the microbe does not produce catalase or significant levels of superoxide dismutase and is ill-equipped to deal with the oxygen radicals produced in the phagolysosome. Minutes after being engulfed by a phagocyte, the cells are dead. The end result of antibody production is a rapid clearing of pathogens in the area of infection. Microbial numbers begin to decrease rapidly and within a few days, the infection and its symptoms dissipate.
In reality, this course of events does not happen in a patient with strep throat. Individuals who are infected with S. pyogenes are given penicillin (or another effective antibiotic), to which the microbe has never become resistant. This is fortunate because some of the antigens present on S. pyogenes are similar to self-antigens on the heart valve and cartilage. In about 1-3% of cases, the development of an immune response against S. pyogenes can lead to rheumatic fever, an autoimmune disease where antibodies directed against the M protein of S. pyogenes attack the heart values and joints. To prevent this serious and undesirable immune reaction, S. pyogenes infections are treated with antibiotics before the body has a chance to mount a vigorous immune response.
Host Response to Viral Pathogens Relies Heavily on T Cells
The response to a viral infection is quite different from that seen in a bacterial infection. Viral infections are intracellular for the most part, while the case we described above was extracellular. This has two implications. First, much of the damage is going to occur inside infected cells. Second, elimination of the infection is going to require destruction of host cells. Therefore, the immune system needs a method of discriminating virally infected cells from healthy cells. Fortunately our bodies have just such a mechanism and it involves the MHC I molecules. In general, cell-mediated immunity is much more important for clearing a viral infection. Figure 16.13 summarizes the hosts response to infection with influenza virus.
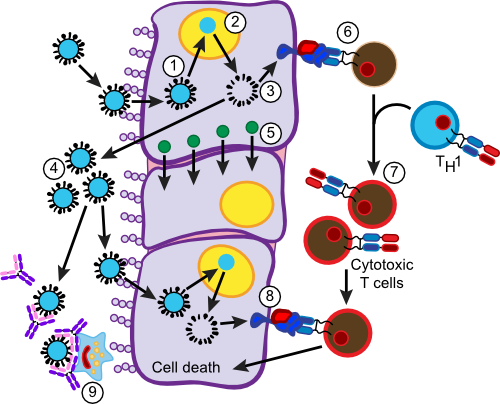
Figure 16.13. Immune Response to Influenza Infection. An immune defense against a viral infection is more dependent on T cells and less dependent on antibodies. Cytotoxic T cells are important in killing virally infected cells. In step (1) influenza virus enters the cell and begins to replicate (2). Viral proteins fill the cytoplasm (3). Most go on to form influenza virus and escape (4). The presence of viral proteins in the cytoplasm also causes the production of α-interferon (5). Some viral proteins are degraded and end up being displayed in MHC I molecules (6). Passing Tc cells will test the MHC I molecule presenting foreign antigen and a fraction will be activated by it. Activated Tc cells are directed to differentiate into cytotoxic T cells by TH1 cells. Activated cytotoxic T cells then attack other virally infected cells displaying viral antigens in the MHC I molecules that the T cells react to. Viral antigens are also presented to B cells producing antibodies in a similar fashion to that described in Section 16.25. These antibodies attack free virus, agglutinating it and making it available for phagocytosis (9).
Influenza virus is a good example to study because it illustrates many of the salient points of viral infection and is also an important human pathogen. However, remember that the specific response of the immune system is dependent upon the particular pathogen. Many of the mechanisms that we describe here come into play in other viral infections, but the exact response is always unique to the particular viral agent.
Influenza is a very contagious disease that easily spreads through respiratory droplets expelled by infected individuals. For this example, imagine that an influenza sufferer has just sneezed on their hand and then opened a door, contaminating the door handle. Another person touching the handle can picked up droplets containing 100,000,000 flu viruses and a subsequent touch can transfer 1,000,000 to the nasal or oral cavity. Some of them then land at the back of the throat. Again, the low pH and unfriendly environment created by the normal flora and host proteins cause the vast majority of the viruses to be inactivated. Those that do survive bind to sialic acid-containing proteins or lipids on the surface of throat epithelial cells and enter by receptor-mediated endocytosis. A drop in the pH of the endosome causes a conformational change in the virus and the release of the eight genomic fragments of influenza virus into the cell. Up until this point, the immune system has no indication that anything is amiss.
Virus begins to replicate and viral proteins accumulate in the infected cell. Some of these proteins are degraded by host cell machinery and the peptide fragments are transported into the endoplasmic reticulum, where they combine with newly synthesized MHC I molecules. The MHC I molecules loaded with foreign viral antigens now find their way to the surface of the cell. The infected cell also begins to produce and secrete α-interferon. This notifies surrounding cells of viral infection and induces them to produce compounds that interfere with viral replication making further infection more difficult.
Virally infected cells begin to release flu virus particles into the surrounding tissues. The presence of viral particles and the death of virally infected cells begin the process of inflammation as described for S. pyogenes infection. This causes the characteristic redness, soreness and swelling in the back of the throat and the induction of fever observed in influenza. At this point, macrophages and dendritic cells take up viruses and viral debris, process them and add to MHC II molecules for presentation to T helper cells. TH1 cells detect the presented antigen and those that match are activated and begin to secrete IL-2, which has several effects on other T and B cells responding to the infection.
Mucous secretion from the intensifying inflammation begins to cause a runny nose and coughing. The release of interferon and IL1 contribute to the aches and fever associated with influenza. As the concentration of virus increases in the body, these symptoms intensify.
Cytotoxic T cells (Tc cells) roaming in the tissues encounter infected cells presenting viral antigens in their MHC I molecules. Those that have TCRs that respond to the antigen are activated to begin clonal expansion and develop into active cytotoxic cells under the influence of the TH1 cells. During the next encounter with a virally infected cell, these cells again recognize the viral antigen being presented in an MHC I molecule using their TCR, but this time, they are activated to attack the cell. The Tc cell binds to the infected cell and begins a destructive cycle. A number of cytokines, including γ-interferon and tumor necrosis factor (TNF), are secreted by the Tc cell. These factors limit viral replication in the target cell and also attract phagocytes to the area. The Tc cells also produce molecules that elicit a form of programmed cell death (apoptosis) in the target cell, essentially telling the cell to kill itself (see section 16.3). Phagocytes that enter the area then clean up any remaining viral debris. As in the case of B cells, some of the Tc cells of the clonal expansion do not differentiate, but remain as memory cells in preparation for the next viral challenge by this viral strain.
Phagocytized virus is also presented to TH2 cells that respond by activating in a manner identical to that described earlier for bacterial infections. Virally infected cells also stimulate B cells that respond by clonal expansion and differentiation into plasma cells, resulting in the formation of antibodies against viral antigens. Antibodies are commonly raised against hemagglutinin and neuraminidase, two proteins on the outer surface of the virus. In most viral infections, antibody is not as important as in bacterial infections, but it does have several consequences. Antibodies bound to virus cause them to agglutinate, precipitate them out of solution and slow their spread through the body. Many effective antibodies block the receptor site of the virus and prevent its attachment to new host cells. These types of antibodies are especially useful in stopping subsequent infections by the same virus. Antibodies attached to virus also assist phagocytes in the efficient uptake of viral particles.
Free virus in the body is eliminated by the action of antibodies and phagocytes and the activated Tc cells destroy any virally infected cells. As the load of virus present in the body decreases, T suppressor cells help the immune response abate. After infection, subsequent attack by this strain of influenza virus is prevented by the action of T and B memory cells.
Key Takeaways
- The response of the immune system to a pathogen is integrated and involves all of the cells and tissues that have been discussed in this chapter.
- This immune response is different, depending upon the type of pathogen encounter, with a major distinction of response occurring based upon whether an infection is extracellular or intracellular.
Quickcheck 16-5
Warning, you must be logged in to be able to have your exam graded. Answer the questions below and if you are a registered user of the site you will see a Grade Exam button. Click it to have your exam graded.