Latest News
- Clues beginning to emerge on asymtomatic SARS-CoV-2 infection
- Back in November of 2020, during the first wave of the COVID-19 pandemic, I was teaching an in-person microbiology laboratory. One of my students had just been home to see his parents, and they all c…
- Read more
- Could there maybe be better uses of genetics and probiotics?
- Professor Meng Dong and his laboratory have created a probiotic that can metabolize alcohol quickly and maybe prevent some of the adverse effects of alcohol consumption. The scientists cloned a highl…
- Read more
- ChatGPT is not the end of essays in education
- The takeover of AI is upon us! AI can now take all our jobs, is the click-bait premise you hear from the news. While I cannot predict the future, I am dubious that AI will play such a dubious role in…
- Read more
- Fighting infections with infections
- Multi-drug-resistant bacterial infections are becoming more of an issue, with 1.2 million people dying of previously treatable bacterial infections. Scientists are frantically searching for new metho…
- Read more
- A tale of two colleges
- COVID-19 at the University of Wisconsin this fall has been pretty much a non-issue. While we are wearing masks, full in-person teaching is happening on campus. Bars, restaurants, and all other busine…
- Read more
( 35982 Reads)
|Learning Objectives
After reading this section, students will be able to...
- Describe the basic structure of an amino acid.
- Explain the peptide bond and how proteins are built.
- Identify the secondary structures that proteins fold into and understand the role of protein motifs in enzyme function.
- Describe tertiary and quaternary structure of proteins.
Proteins and peptides (small proteins) are essential to the cell and serve two major functions. Many proteins are enzymes that catalyze almost all biological reactions in a living organism. Other proteins perform a structural role for the cell - either in the cell wall, the cell membrane or in the cytoplasm. In this section, we will look at the basic structural elements shared by all proteins.
Structure of Amino Acids
Proteins are polymers of amino acids. Amino acids, with rare exception, contain an α carbon that is connected to an amino (NH3 ) group, a carboxyl group (COOH), and a variable side group (R). Figure 2.19 shows this generic amino acid structure. The side group gives each amino acid its distinctive properties and helps dictate the protein's folding.
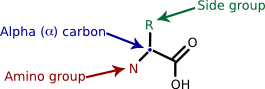
Figure 2.19. A generalized amino acid. Each amino acid contains a carboxyl group, an amino group and a variable side group (R). These all connect to a central carbon, termed the α-carbon, identified by the arrow.
The primary structure of proteins is the amino acid sequence
As with nucleic acids, primary structure refers to the ordered sequence of the different amino acids in a protein. The carboxyl group and the amino group of amino acids are reactive. As shown in Figure 2.20, cells synthesize proteins by attaching the carboxyl group of one amino acid to the amine group of the next, with polymerization taking place at the ribosome. These covalent bonds in proteins are termed peptide bonds. Since each amino acid has a carboxyl group and an amino group, hundreds or thousands of amino acids can be linked together.
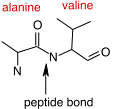
Figure 2.20. A peptide bond. The peptide bond between an alanine residue and a valine residue is identified by the arrow. Peptide bonds can form between any two of the 20 amino acids and link the carboxyl group of one amino acid and the amine group of the next.
There are 20 common amino acids found in proteins, and these amino acids can be roughly classified into three groups: polar, non-polar, and charged. Polar and charged amino acids are hydrophilic and are often found on the surface of a protein, interacting with the surrounding water. In contrast, non-polar (or hydrophobic) amino acids avoid water. While this categorization is adequate for most purposes, you should recognize that it is a bit simplistic. For example, arginine does have a charged hydrophilic group at one end, but the -CH2 - backbone that makes up most of the amino acid is quite hydrophobic. Figure 2.21 shows the chemical structure of all 20 common amino acids.
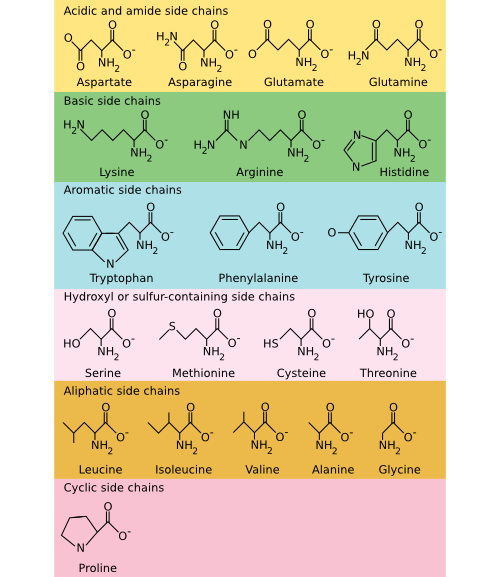
Figure 2.21. The common amino acids. The amino acids are organized into their chemical type and characteristics. Acidic and basic amino acids carry a charge, amino acids with a sulfur or hydroxyl component are polar, and aliphatic and aromatic amino acids are non-polar.
Secondary structure is the local geometry of the protein
Ribosomes, and the rest of the translation machinery, link amino acids together to form peptides and proteins into polymers that range from 10 to 10,000 residues in length. During and after protein synthesis, the residues of the primary sequence dictate how the protein folds. The simplest aspect of protein folding is termed its secondary structure, which refers to the geometry of the local polypeptide chain with respect to its immediate neighbors. While the primary sequence of amino acids dictates protein folding, predicting the overall structure from the primary sequence remains one of biology's most important unsolved problems. Nevertheless, the major determinants of this final structure are hydrophobic interactions. The protein must hide hydrophobic amino acids from the water interface during protein folding by burying them in the interior. This burying defines the protein core, which then influences the immediate structure around it and greatly affects the protein's overall structure.
Common Secondary Structures
Peptide bonds between adjacent amino acids can rotate and twist to allow a large number of interactions. Still, two local organization schemes, the α helix and the β sheet, are found in many proteins. Their prevalence is prevalent because they happen to form particularly energetically favorable structures. Numerous hydrogen bonding opportunities and hydrophobic interactions between nearby amino acids in the protein drive the formation of these structures. The α helix resembles a ribbon of adjacent amino acids wrapped around a tube to form a staircase-like structure. Figure 2.22 shows different representations of an α helix. This structure is very stable, yet flexible and is often present in parts of a protein that may need to bend or move.
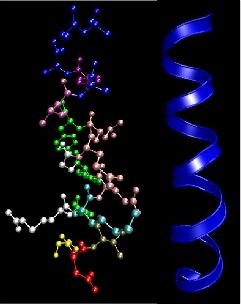
Figure 2.22. The α helix motif in proteins. This figure shows two common depictions of an α-helix, an extremely common and important motif in proteins. The ball-and-stick depiction on the left shows the various side chains, but the helical nature of the structure is not so obvious. The ribbon representation on the right only traces the backbone of the peptide bonds and overemphasizes the symmetry a bit, but shows the helical nature of the structure.
In the β sheet, two protein chains (perhaps different segments of the same protein) align themselves in a planar structure such that hydrogen bonds can form between facing amino acids in each sheet. Figure 2.23 shows different representations of this structure. The β sheet is different from the α helix in that it can involve amino acids from different sections of the protein, which come together to form this structure. Also, the structure tends to be rigid and less flexible than the α helix.
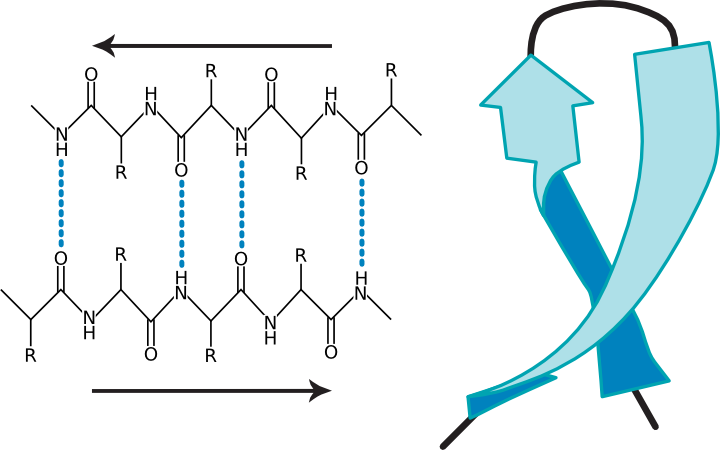
Figure 2.23. Two views of a β-sheet. (a) A diagram of a β sheet showing hydrogen bonding between protein strands (b) A ribbon representation of a β-sheet.
Tertiary structure is the 3D structure of individual polypeptides
The entire protein chain will fold into a series of α helices and β sheets connected by strings of amino acids that form other structures. The overall structure of the folded protein is termed its tertiary structure.
As noted before, the most important stabilizing force in proteins is burying hydrophobic residues from the surrounding water. However, other chemical features are important in creating and stabilizing tertiary structures. These include hydrogen bonds, ionic interactions, and sulfhydryl bonds. Section 2-1 described hydrogen bonds and ionic interactions. Sulfhydryl linkages are unique to proteins and are covalent bonds between cysteine groups. Cysteine is a unique amino acid in that it has a sulfur group at the end of its variable side group that is available for binding to other groups. Often in proteins, nearby sulfhydryl groups on cysteines form a covalent bond, and these are often crucial for the stabilization of the mature protein for it to perform its function. Figure 2.24 shows some representations of sulfhydryl bonds in proteins.
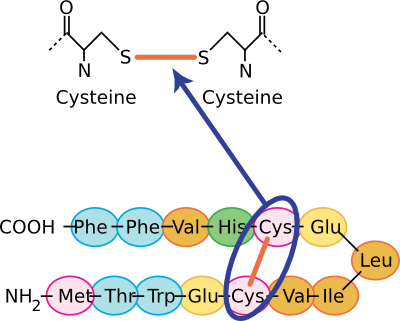
Figure 2.24. The chemical structure of a sulfhydryl bond. Several views of sulfhydryl linkages are shown. In the top panel, the covalent sulfhydryl linkage between two cysteine residues is shown. In the bottom panel, this sort of Cys-Cys bond is shown in the context of a section of a protein.
All of these forces combine to fold a protein into its final tertiary structure. Figure 2.25 shows the complete structure of ribulose bisphosphate carboxylase (RubisCo), one of the most abundant enzymes on this planet. This enzyme takes energy, obtained most often from the sun, and uses it to convert carbon dioxide into carbohydrate. Its abundance reflects the fact that virtually all photosynthetic organisms, from bacteria to plants, use this enzyme to produce much of the cell's carbon. Note the α helices throughout the protein and the β sheet near the bottom. These structures help to hold the protein in its proper conformation so that it can carry out its enzymatic activity.
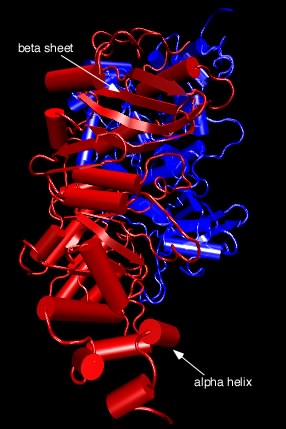
Figure 2.25. Tertiary structure of ribulose bisphosphate carboxylase.. The figure shows the structure of ribulose bisphosphate carboxylase, typically abbreviated Rubisco, from the bacterium Rhodospirillum rubrum . As described in the text, this complex structure has alpha helices (shown as cylinders) and beta sheets (shown as the flat ribbons) throughout.
Now, we have talked about certain secondary structures that recur in many proteins, but it also happens that there are conserved tertiary structures as well. In other words, there are regions of proteins, called motifs or domains, that are structurally identical in two or more different proteins. Figure 2.26 shows some examples of protein motifs. By "structurally identical," we mean that they have similarly sized α helices and β sheets organized in a similar overall way. Interestingly, these domains do not always have identical primary structures. Instead, it appears that a variety of different sequences can fold to give the same overall tertiary form. However, they will often have conserved amino acids in them at critical positions. These motifs often have a specific function. For example, there is a widespread motif that proteins use for binding ATP. A second class of examples would be the set of motifs that help bind DNA. Through genetic changes, these motifs have been grafted onto other protein regions so that the same ATP-binding (or the DNA-binding) domain performs a similar function for proteins with completely different overall activities. Of course, the unique functions are defined by those portions of the protein that are not in common.
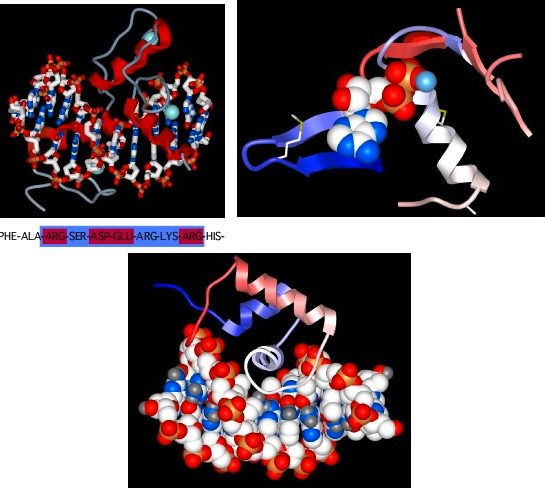
Figure 2.26. Some common protein motifs. Three examples of protein motifs are shown. The top left panel shows two zinc-finger motifs (the red helices and thin gray lines), each bound to a Zinc atom shown in pale blue. This is a common motif found in DNA-binding proteins and the interaction is shown here (the ball-and-stick figure running from left to right in this panel is a section of DNA). Note that the Zinc does not interact with the DNA, but instead stabilizes the protein motif to allow it to form the precise shape to bind to DNA. The top right panel shows an ATP-binding domain, with the ATP displayed as the space-filling model and the protein shown in a ribbon format. The bottom panel shows another common DNA-binding motif, termed the helix-turn-helix. Here the DNA is shown in space-filling form and the two helices after which the structure is named are shown in red and white (the latter is shown end-on and is interacting with the DNA).
From this description, you think of proteins as a collection of protein pieces, each of which serves a function and has an evolutionary history. We refer to these as functional domains. The enzyme pyruvate kinase, which generates ATP by removing it from phosphoenolpyruvate, has three domains, termed A, B, and C. The A domain is responsible for binding substrate (phosphoenolpyruvate and ADP) while the C domain serves a regulatory role, binding fructose-6-bisphosphate and stimulating activity.
Up to this point, we have talked as if proteins are static, rigid structures, but this is certainly wrong. Instead, most proteins are flexible, dynamic structures that respond to the conditions around them. Movement is typically essential to their function, especially for enzymes that need to grab a substrate, react with it, and release product. This movement can be just a few tenths of an angstrom (10 -10 meters), while in other proteins, this movement can be quite large. This shift obviously changes the tertiary structure of that protein. Thus, when we talk about tertiary structures, typically determined by crystallography, we refer to only one of the possible structures that the protein can exist in while performing its biological function.
Quaternary structure is the total complex of a functional protein
Many proteins are actually complexes of several polypeptides. The quaternary structure of a protein is the arrangement of more than a single polypeptide into a collection. Protein complexes might contain two or more copies of the same protein, or they may consist of any number of different polypeptides in various ratios. Such complexes are certainly not random but reflect precise interactions among the protein subunits based on the same sorts of interactions (e.g., hydrophobic, hydrogen bonds, etc.) described above.
Figure 2.27 shows the catabolite activator protein, an example of a protein containing identical subunits. In this case, it has two subunits, termed a dimer. Each subunit has a cyclic adenosine monophosphate molecule bound that activates the protein. In the active state, this protein binds DNA and activates various genes in the cell.
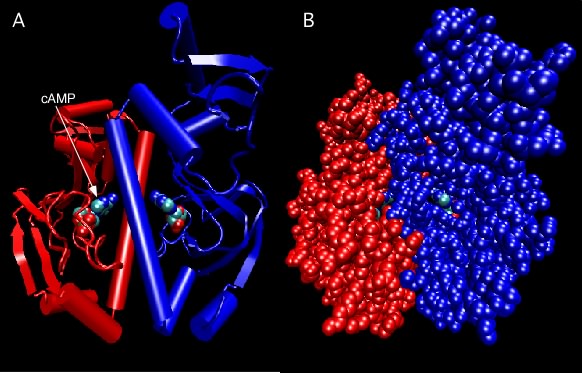
Figure 2.27. Catabolite Activator Protein structure. Two depictions of the catabolite activator protein (CAP) of E. coli are shown. The left panel shows a ribbon depiction, with the two identical subunits shown in red and blue. CAP senses the presence of cyclic AMP (cAMP) in the cell and a molecule of cAMP bound to each monomer as indicated. The dimer is actually symmetrical, but that is not obvious here because the model is rotated slightly. The right panel shows a ball-and-stick representation of the same dimer and gives a better sense of the overall shape. Note that the bound cAMP molecules are buried within the protein (so how do they ever come and go?).
Dinitrogenase is an example of a protein containing non-identical subunits. As shown in Figure 2.28, the active protein has two copies of one protein (termed α) and two copies of a second protein (termed β), and therefore is also referred to as an α 2 β 2 tetramer. This protein is responsible for the reduction of N2 gas to ammonia and is critical to global nitrogen cycling. The importance of the quaternary structure of dinitrogenase is only partially clear. The α subunits contain the metal cluster where N2 is reduced (or "fixed") and the β subunit helps form another metal center that helps transfer electrons to that active site. We can therefore understand why there might need to be an αβ dimer. However, it is unclear why there are two such αβ dimers hooked together in the tetramer found in nature. There is, for example, no apparent communication between the two active sites.
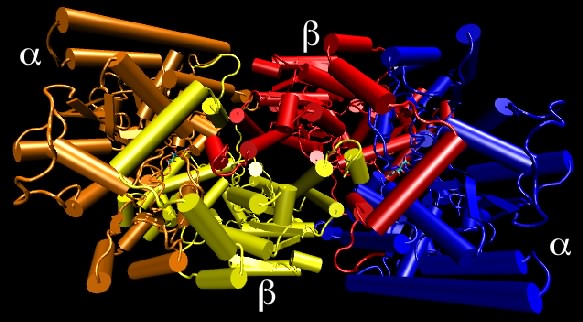
Figure 2.28. Nitrogenase structure. Nitrogenase is made up of four protein chains, with two copies each of different proteins, termed the α and β subunits. Though not apparent in this view, the two α subunits are identical to each other, as are the two β subunits. The quaternary structure is referred to as an α2β2 tetramer.
Glycoproteins are proteins with attached sugars that are typically important for the proper function of the protein. In most cases, enzymes add the sugars to the protein after translation. Proteins that encounter the outside environment of the cell are sometimes glycosylated to stabilize them against attack from degradative enzymes and destructive physical forces.
Key Takeaways
- Amino acids are organic acids with an amine group attached to the α carbon. Amino acids are distinguished by their side group that dictates their properties.
- Proteins contain 20 different amino acids that are linked by peptide bonds to form long polymers. Amino acids can be classified into three groups, polar, non-polar, and charged.
- The chain of amino acids dictates how a protein folds. The major determinant of protein folding is hydrophobic interactions. The local geometry of a protein is its secondary structure. Two common secondary structures are the α helix and the β sheet.
- Tertiary structure is stabilized by hydrophobic interactions, hydrogen bonds, ionic interactions, and sulfhydryl bonds.
- Conserved tertiary structures are called motifs. Many common motifs have been discovered. Some examples are ATP-binding and DNA-binding motifs.
- Proteins are flexible, dynamic structures that respond to their surroundings.
Quickcheck 2-6
Warning, you must be logged in to be able to have your exam graded. Answer the questions below and if you are a registered user of the site you will see a Grade Exam button. Click it to have your exam graded.