Latest News
- Clues beginning to emerge on asymtomatic SARS-CoV-2 infection
- Back in November of 2020, during the first wave of the COVID-19 pandemic, I was teaching an in-person microbiology laboratory. One of my students had just been home to see his parents, and they all c…
- Read more
- Could there maybe be better uses of genetics and probiotics?
- Professor Meng Dong and his laboratory have created a probiotic that can metabolize alcohol quickly and maybe prevent some of the adverse effects of alcohol consumption. The scientists cloned a highl…
- Read more
- ChatGPT is not the end of essays in education
- The takeover of AI is upon us! AI can now take all our jobs, is the click-bait premise you hear from the news. While I cannot predict the future, I am dubious that AI will play such a dubious role in…
- Read more
- Fighting infections with infections
- Multi-drug-resistant bacterial infections are becoming more of an issue, with 1.2 million people dying of previously treatable bacterial infections. Scientists are frantically searching for new metho…
- Read more
- A tale of two colleges
- COVID-19 at the University of Wisconsin this fall has been pretty much a non-issue. While we are wearing masks, full in-person teaching is happening on campus. Bars, restaurants, and all other busine…
- Read more
( 32817 Reads)
Many modern techniques for detecting the presence of microorganisms and determining the sequence of their DNA depend on molecular biology techniques that first began to appear in the middle of the 1970's. In this section we give a somewhat detailed explanation of common methodologies that were used in part to generate much of the information we have talked about throughout this textbook. Of course the techniques talked about here are not comprehensive, but they do cover some of the more common (and oft mentioned) procedures of microbial molecular biology in use today.
Gel electophoresis
It is often very useful to separate different types of biological molecules so we can detect, analyze or even isolate them. When molecules with a charge are placed in an electric field and a current is applied, they move. One could therefore put electrodes on opposite sides of a beaker full of cell extract and the positively charged molecules would move to the negative electrode and negatively charged molecules would move to the positive electrode. This would not be very useful, because you could not easily tell where any molecules were in that solution nor could easily remove them without mixing. Molecular biologists therefore create electric fields in semi-solid material called gels. These can be made from a variety of things, from seaweed extract (agar and agarose) to chemically synthesized systems (acrylamide), but they have some similar properties. In all cases, they are loose structures that are full of water, but they differ in how dense the structures are. In other words, the size of the holes through the material can vary. If one creates a gel Figure 28.26, puts opposite electrodes at each end and puts some cell material in a little well in the center, the different molecules will migrate toward one electrode or another when the current is turned on. The rate at which they migrate will depend on their size and charge. Very charged molecules are strongly attracted to one electrode and move faster (all other things being equal) than less charged molecules. However, the molecules also need to wiggle through the holes in the gel, so small molecules move faster than larger ones (again, if all other factors are equal). Remarkably, the speed of migration is highly reproducible, so that all fragments of the same size and charge migrate at the same rate within a few percent.

Figure 30.26. An agarose gel. Pictured here is an agarose gel before it is run. Note the lanes at the top of the gel where sample can be loaded.
A critical feature of gels is that, unlike the case with the beaker above, when one turns off the current, the molecules not only stop moving, but one can find out where they are because they are fairly well trapped in the gel. Gels can even be dried down and saved. You can also determine the positions of specific molecules identified by various methods. But how do you know where these molecules are in the gel? There are several different approaches, depending on the nature of the molecules that are in the gel. Nucleic acids can be detected by stains that bind to only the nucleic acid and not to the gel, or by using radioactively labeled nucleic acid and then exposing the dried gel to film, which is exposed near the radioactivity (termed autoradiography). For proteins, detection of all proteins in the gel can also be performed with general stains or radioactive labeling.
It is even possible to detect a specific protein or a specfic nucleic acid in a gel and we will describe these methods in greater detail below.
Restriction Enzymes
Restriction enzymes evolved in many species of prokaryotes as a mechanism to protect themselves from foreign DNA that might enter the cell (see T4 in Chapter 13). Some of this foreign DNA might be that of bacterial viruses, but there are other sorts of selfish DNA molecules that a cell might want to protect itself from. The typical solution was an enzyme that would recognize a specific DNA sequence, typically 4 or 6 base pairs in length, and then cut both DNA strands whenever that sequence was found. Such a double-strand break destroys the ability of most DNA molecules to replicate and therefore would protect the host cell from entering DNA molecules. But how does the cell prevent its restriction enzyme from cutting the same sequences in its own DNA? The solution is that cells produce an enzyme that methylates the identical DNA sequence on both DNA strands, so that the restriction enzyme fails to recognize and cut it. So why doesn't the methylation system also protect the foreign DNA? That is because the foreign DNA will enter the cell in an unmethylated form, which is a very good substrate for the restriction enzyme and a poor one for the methylase. In other words, in most cases the restriction enzyme gets to the foreign DNA first, before the methylase can modify it. But that brings up yet another question: concerning that host DNA, how does the methylase beat the restriction enzyme? In this case the host DNA is normally methylated on both strands, but after replication, only the old strand is methylated. This hemi-methylated site is a very good substrate for the methylase (so the site is quickly converted to a fully methylated form), but is a terrible substrate for the restriction enzyme.
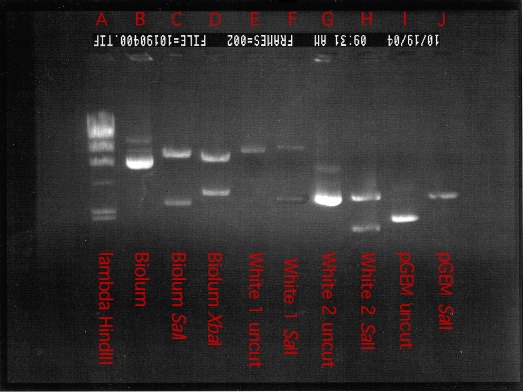
Figure 30.27. A finished agarose gel. Various plasmids isolated as part of a cloning experiment were run out on an agarose gel. (lambda HindIII) phage lambda DNA digested with the restriction enzyme HindIII. (A) the plasmid pGEM3Z from Promega corporation with the bioluminescent genes from Vibro fischeri cloned into it. (B) Plasmid in A digested with SalI. Note that he insert is digested out of the plasmid and is about 10 kB long. (C) Plasmid in A digested with XbaI. In this case the insert has an XbaI site and is cut. The plasmid also has one XbaI site. (D). (E-H) Various cloned inserts from Vibrio fischer. (I) plasmid pGEM3Z. (J) plasmid pGEM3Z cut with SalI.
Our use of restriction enzymes has little to do with their biology. As you might guess, if different organisms are going to use these systems for identifying foreign DNA, then each species needs to have different restriction enzyme target sites than most other organisms. As a consequence, there are a large number such enzymes, typically from different prokaryotic species, that have completely different target sequence. Companies over-express and purify these different enzymes of known target sequence and sell them, allowing the experimenter to cut their DNA at a variety of specific sequences. This is valuable in itself, since it can give you a lot of DNA of precise size and you can, for example, cut a plasmid preparation and run the fragments on an agarose gel, Figure 28-27, and isolate a particular DNA fragment. However the greatest utility of these enzymes has come from another property; they typically make staggered cuts at the site. This staggering means that there will be a 1-4 nucleotide region of single stranded DNA at every newly created end. Moreover, the single-stranded region of one end will be able to base-pair with the single-stranded region of any other DNA end produced by the same enzyme. This base pairing of a few bases is not strong, but it is good enough to allow DNA ligase, an enzyme that seals nicks in DNA, to fix both nicks and form a normal piece of double-stranded DNA. This has been critical for much of molecular biology because it allowed the scientist to cut-and-paste different DNA fragments together in a precise way. The increased use of PCR has made some of the uses of restriction enzymes obsolete, since PCR can be used to fuse any two sequences together and does not depend on the existence of specific sequences at desired positions (see below).
The restriction enzymes referred to above are ones that have relatively small recognition sequences of 4-6 bp, so the number of sites with such a sequence is fairly large in a piece of DNA as large as a chromosome. However, there are some enzyme that recognize longer sequences (8 bp) and probability dictates that such sequences are extremely rare. As a consequence, cutting an entire chromosome with such enzymes often gives a few to a few dozen discrete bands. These very large fragments can be very useful in trying to organize DNA sequence data (see below) into a pattern that represents the circular chromosome.
Cloning
Cloning has a variety of meanings that are only distantly related. In general, it means to produce many identical copies of something. For a prokaryotic molecular biologist, cloning has meant the ability to produce large amounts of a single DNA sequence. For a number of years, this was accomplished by using restriction enzymes to move the desired piece of DNA onto a vector like a plasmid or the genome of a bacteriophage. In each case, one can grow up cells with many copies of the vector, isolate the plasmid or phage, and then remove the desired piece of DNA from the vector using restriction enzymes. The different fragments were separated from each other on a gel. It was thus possible to make as many copies of the DNA as needed for experiments and one would then have cloned that specific DNA sequence. This process is diagrammed in Figure 28.28. Today, one typically obtains large amounts of a specific sequence more directly by PCR, which can amplify large amounts of DNA defined by the choice of two DNA primers.
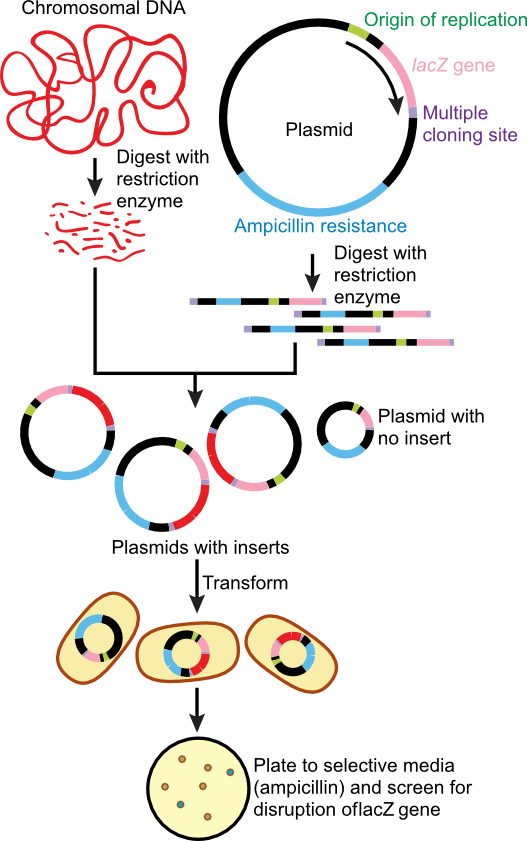
Figure 30.28. Cloning of target DNA. In the classic method of cloning, often called shot-gun cloning, purified target DNA is digested with the same restriction enzyme as a plasmid that will carry the DNA. They are then mixed together and the overhang of bases, created by the restriction enzyme digestion, hybridize. These newly formed plasmids, containing target DNA, are covalently closed using DNA ligase and transformed into a suitable host. Since transformation is an infrequent even, the plasmid is then selected for on solid medium containing a drug resistance the plasmid carries. In this case ampicillin.
As you might know from newspaper and movies, however, cloning has a rather different meaning: the production of multiple copies of an identical animal. Typically this is done by teasing apart the few cells of an early-stage fertilized egg, and then using each of these to create new and genetically identical individuals. Alternatively, cell nuclei can be extracted from certain cells of an adult animal and put in an environment (an egg) where again they can develop to a new organism. While there are certainly a number of ethical issues involved in this, particularly in the case of humans, you'll realize that this is roughly what prokaryotes do all the time.
Transformation
Transformation is the method that allows the introduction of bare DNA into a cell and naturally occurring transformation was covered in Chapter 12. Transformation has two hurdles to overcome: first, the DNA must be purified and this can be either plasmid or chromosome, linear or circular; and second, competent cells must be generated. Competent cells are those that can accept DNA. In the case of E. coli, there are a variety of recipes for performing this, but typically they involve multivalent cations and some time of incubation at low temperature, whereupon, a fraction of the cells (fewer than 10%) become competent to take up DNA. Somehow the process leads to the formation of dozens of "channels" per cell that can take up DNA. A number of other gram-negative bacteria have been successfully transformed with variations on this approach.
Gram-positive bacteria have to be treated differently, because of the differences in their cell wall. They can be incubated with degradative enzymes to remove the peptidoglycan layer and thus form protoplasts. When these latter cell forms are incubated with DNA and polyethylene glycol, one obtains cell fusion and concomitant DNA uptake. In both of these examples, if the DNA is linear, it tends to be very sensitive to nucleases so that transformation is most efficient when it involves the use of covalently closed circular DNA. Alternatively, nuclease-deficient cells can be used to improve transformation.
Finally, the technique of electroporation is becoming commonly used with bacteria. It essentially involves creating transient pores in the cells using electric current and can be used for both cell fusion and incorporation of DNA. When optimized for a particular organism, electroporation seems to be good for at least an order of magnitude increase in the frequency of transformants when compared to optimized transformation methods and in some species is the only efficient way of getting DNA into cells.
DNA Sequencing
DNA sequencing has gone through huge changes in the past 30 years. In the late 1960's it took a graduate student 4 years to sequence 12 bases that form the sticky ends of phage lambda and now millions of bases are sequenced daily around the world. Continuous innovation keeps changing the details of the way DNA is sequenced, but the method is still based on the idea of running reactions that distinguish the four different types of bases and causing random errors in the process so that an entire spectrum of different sized fragments is created.
Currently, most sequencing reactions rely on synthesis of DNA using DNA polymerase from thermophilic organisms. In a manner similar to PCR, the reaction contains a template to be sequenced, a primer and the four nucleoside triphosphates. Note however, that there is only one primer, not two as in PCR. To this reaction is also added a small amount of the four nucleoside triphosphates where the 3'-OH has been removed - they are referred to as di-deoxy nucleotides. Incorporation of one of these nucleotides causes the chain to terminate, since there is no 3'-OH for the DNA polymerase to act upon. The low concentration of di-deoxy nucleotides means that there is only a small chance that the chain terminates at any step upon the DNA template and thus a range of differently sized fragments are generated representing termination at each base along the sequence. Each fragment terminates with one of the four di-deoxy nucleotides and the specific one depends upon the sequence of the template at that position. To make identification easy, each di-deoxy nucleotide (ddATP, ddCTP, ddGTP and ddTTP) is labeled with a different colored fluorescent dye and thus it is possible to determine the ending nucleotide on each sequence. After running the sequencing reaction, the reaction mix is separated based on fragment length using electrophoresis either through a gel or using a capillary system. Fragments coming out the end of the gel are read using a laser and the base pair at that position is recorded. The Dolan DNA Learning Center has a nice animation on the process of DNA sequencing.
Mutated forms of DNA polymerase increase the efficiency of incorporation of these labeled nucleotides. These forms of DNA polymerase are also heat stable, thus allowing heating of the reaction mix to denature DNA and allowing several cycles of DNA replication from the same template (thus cutting down on the amount of DNA required). In the future the method used is sure to change again and become even more efficient, but the above discussion gives you an idea of the type of reactions run today.
Genomics
Genomics is a rather general term for the use of genome sequences to do something useful and was covered in some detail in Chapter 12. Genomics requires the availability of large amounts of DNA sequence information. One application of genomics, sometimes called functional genomics, is the analysis of the amount of gene expression in a cell under different conditions. This very powerful method is covered below in the section called DNA arrays. Indeed, merely using a sequence to identify an interesting gene and clone it by PCR is a form of genomics.
A rather different form of genomics involves the computer analysis of the sequence information, which is often termed bioinformatics. One routine approach is to compare the sequence of genes for which we already know the protein function and look for sequences that are highly conserved. The logic is that random mutation will change many parts of the gene, but the parts of a protein that are essential for its function cannot be changed without damaging that function. Conserved residues can be found in such sequence comparison and the predicted important residues tested experimentally. Another common use is to start with a gene of unknown function and ask what other genes (of known function) are similar to it. The logic here is that if genes are sequentially similar at all, then they must be evolutionarily related. If they are related, then it is highly likely that the product of the unknown gene will have a function similar to that of the known gene. Obviously, the greater the gene similarity, the more likely it is that the protein function will be the same. Finally, one can compare the presence or absence of genes in entire genomes. For example, people have hypothesized that pathogens should have genes that allow them to infect a host. They further supposed that similar genes should be found in at least some other pathogens, but that you should never find them in non-pathogens (because non-pathogens would have no use for them). They therefore looked for genes that satisfied these criteria and many of them have already been shown by other means to be involved in virulence. This suggests that the other genes, found only in pathogens but of unknown function, might also be involved in pathogenicity.
The discussion so far has been about how one sequences DNA, but not how one actually sequences all the DNA of the cell to determine the genome sequence. Initially this was done by cutting the chromosome into small pieces, using restriction enzymes and plasmids, and then sequencing each of those. When you lined up the sequences of each portion, you could recreate the entire chromosomal sequence. However, it became clear to Craig Ventnor that there was a faster way. He realized that you could simply clone lots of random pieces of chromosomal DNA and sequence them all, and you do it in such a way that you effectively sequence every region about ten times. Importantly, however, the random pieces are all different, so that the end points of each sequenced region will overlap with sequence from other clones, Figure 28.29. You then throw all the information into a computer and it is programmed to recognize these overlaps and therefore guess which sequences are next to each other in the genome. This usually allows one to come fairly close to completely solving the entire sequence. Sometimes there are a few gaps, so that the larger sections of completed sequence are connected to each other by other methods, such as the cloning and sequencing of very large restriction fragments described earlier.
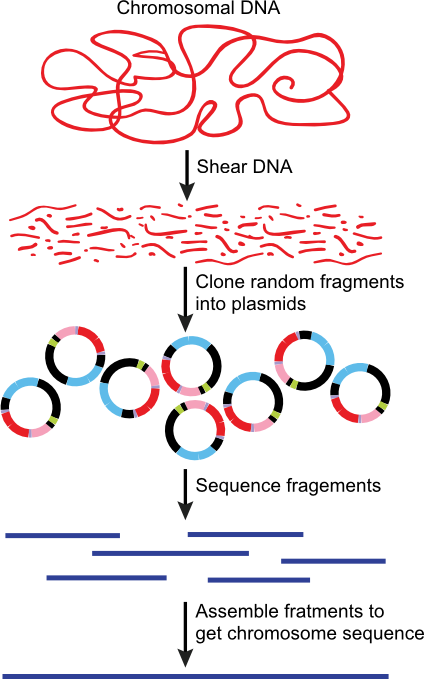
Figure 30.29. Genome sequencing using random fragments. By shearing the DNA, cloning it and sequencing all the fragments, the entire genome of a microbes can be rapidly sequencing. This method can be thought of as creating a giant jigsaw puzzle of DNA fragments. A computer is then used to help assemble the various pieces of sequenced DNA into a completed genome.
Detecting specific nucleic acids or proteins
As briefly mentioned in the section on stains in this chapter, the two strands of a complementary DNA duplex are able to hydrogen bond to each other and form a rather stable complex. A complex can also be formed between two strands that form a less-than-perfect complex, but such imperfect complexes are less stable than perfect ones. These properties form the basis of a number of highly valuable methods in molecular biology.
Let's say that you want to know if a particular DNA sequence is present in the microbe you are working on (whose genome has obviously not been sequence, or you would already know this information). You can take the DNA from your organism and digest it with a certain restriction enzyme, so that every possible cut is made in that DNA. The DNA is then run on an agarose gel. Now because of the phosphate backbone, all nucleic acid molecules have the identical charge per unit length, so the only reason that they might migrate differently is if they have a different length. Therefore in an agarose gel, the large fragments migrate much less than the small ones. After turning off the current, one can stain the DNA in the gel and see a pattern of many different bands, Go back and look at Figure 28.27 for an example).
But how can you ask if the DNA region you are interested in is present in that mess of bands? You take the gel above and transfer the DNA directly to a piece of membrane, to which it sticks in the identical pattern that it existed in the gel as shown in Figure 28.30. . Most of these methods begin with transferring the contents that have been separated by the gel onto some type of membrane that will hold either the nucleic acid or the protein. The finished gel is laid onto a membrane and then sandwiched between two pieces of filter paper. This is then hung in a buffer and a second eletric field is applied that causes the protein or nucleic acid to migrate out of the gel and get stuck on the membrane. This method is called a Southern blot (DNA), a Northern blot (RNA) or a Western blot (protein). This technique was first pioneered by the British biologist Edwin Southern, and it was named after him. Subsequent scientists who worked out conditions for RNA and protein had a bit of fun by naming them in reference to Southern.
Once your molecule of interest is blotted to a membrane, a detection reaction is then run. For detection of specific proteins, antibodies are added to the blot that bind only to the protein of interest. These antibodies have either an attached dye or some other feature that allows the detection of their position and therefore of the target protein.
For nucleic acids, you take a small fragment of DNA for the region that you care about and you tag it in some way - maybe with radioactivity or maybe with certain chemicals - and let this wash over the membrane. The tagged fragment stick tightly, or hybridize, when it finds a complementary strand that is perfect, and sticks weakly where there is a complementary strand that is only somewhat similar. Then you wash off the fragments that have not stuck. By washing under conditions of higher temperature or with formamide, termed high-stringency conditions, you can demand that only perfect or near perfect matches stay on the filter. There might be cases, however, where you want to also see some imperfect matches as well, so lower stringency conditions are used. Finally, you detect the location of the tagged fragment (and therefore the position of the complementing piece). If the tag was radioactive, the detection might use film or a special machine, but other tags have their own detection methods. The position and strength of the signal can be used to determine the fragment size, the abundance and the degree of match to the tagged fragment. Figure 28.30 is a video of the Southern Blot technique
Figure 30.30. The Southern Blot Technique. This is a method of detecting certain DNA fragments in a system.
DNA Arrays
With the advent of genome sequencing and microtechniques borrowed from the computer industry, it has become possible to use hybridization in a rather different way. Scientists have taken the genome sequence of organisms and used PCR or chemical synthesis to make a copy of regions of the chromosome encompassing every single gene, one at a time. These separate amplified fragments are then stuck to a glass or plastic surface in a precise way. This is called a gene array and represents the entire genome of the organism. Now if one took the same sort of tagged DNA fragment described immediately above and allowed it to hybridize to this surface, washed off the poorly hybridizing material and then detected where it remained, you would see which sequences were similar or identical to the tagged fragment. Now if you think about it for a bit, this is not very useful, since you must already know all the sequences involved, so you could have done the same thing by computer. However, this gives you a notion of the method; let's now describe a more interesting use of such an array.
What if we took the microbe for which we have the array and grew it under two different conditions. Let's say that the microbe is a pathogen that is known to turn on genes for virulence in humans whenever the temperature is raised to 37 °C (because the microbe thinks it is in a human body). Now you would certainly like to know which genes were involved in virulence, since you could try to figure out how to interfere with the function of the proteins made from those genes. You therefore grow the microbe at 25 °C (where no virulence genes are expressed) and at 37 °C and you isolate the RNA. For technical reasons, the RNA molecules are reversed transcribed into DNA that is chemically tagged with two different colors for the two culture conditions. The tagged DNA sample from each culture is then hybridized to a separate DNA array, washed and the resultant patterns of hybridization are compared. Intense spots mean that the gene was being heavily expressed, while the absence of a detectable spots means that it was not expressed. An intense spot seen from cultures at both temperatures means that the gene is always heavily expressed. However, one looks for those spots that are much more intense at 37 °C than at 25 °C, as many of these will identify genes whose products are involved in virulence. Figure 28.31 you will find a nice video of the use of a DNA array
Figure 30.31. DNA arrays. DNA arrays are another method of detecting the presence of DNA or RNA fragments in a system. The advantage of a DNA array is that you can detect many with one experiment.
As with any technique, especially a technically involved one such as this, there are a bunch of critical little details that are important for proper interpretation of the results. You really cannot completely remove all of the probe sticking to inappropriate places, so there is a background of signals all over the array. This low background is not a problem for genes that are expressed at high level, since they are well above that background. However, there are important genes whose expression is never very high, such as genes that encode regulatory proteins. The level of mRNA accumulation of such genes might be only a bit above the background of the array, making it difficult to make strong conclusions about them, at least in a quantitative way. Other such technical matters mean that while this method is powerful already, there is still room for improvement.
At its best, this approach gives an unprecedented insight into the physiology of the organism at the moment in time when the mRNA was extracted. It is, in a sense, a description of what the microbe is thinking about at that time.