Latest News
- Clues beginning to emerge on asymtomatic SARS-CoV-2 infection
- Back in November of 2020, during the first wave of the COVID-19 pandemic, I was teaching an in-person microbiology laboratory. One of my students had just been home to see his parents, and they all c…
- Read more
- Could there maybe be better uses of genetics and probiotics?
- Professor Meng Dong and his laboratory have created a probiotic that can metabolize alcohol quickly and maybe prevent some of the adverse effects of alcohol consumption. The scientists cloned a highl…
- Read more
- ChatGPT is not the end of essays in education
- The takeover of AI is upon us! AI can now take all our jobs, is the click-bait premise you hear from the news. While I cannot predict the future, I am dubious that AI will play such a dubious role in…
- Read more
- Fighting infections with infections
- Multi-drug-resistant bacterial infections are becoming more of an issue, with 1.2 million people dying of previously treatable bacterial infections. Scientists are frantically searching for new metho…
- Read more
- A tale of two colleges
- COVID-19 at the University of Wisconsin this fall has been pretty much a non-issue. While we are wearing masks, full in-person teaching is happening on campus. Bars, restaurants, and all other busine…
- Read more
( 67658 Reads)
|There are many different reasons one might need to known the population of microorganisms in a given sample. For example, determining the rate at which a microbe is killed by UV light requires analysis of the number of viable cells before, and at various times during, UV exposure. In other experiments, it is important to know that you have the right density or growth stage to use, such as required for transforming a plasmid constructed in vitro into an E. coli strain. Assessment of bacterial populations in applied industries is also important. Many food-processing plants measure the level and type of microorganisms present in their food by doing counts on selective medium. Also, sewage treatment plants routinely sample and count the microbes present in their treatment systems to insure the correct type and numbers of bacteria are present. The microbial count can be determined using a wide array of techniques. Note that these assays all require somewhat different information and work over different time frames, as explained below. In this section we summarize some of the more common methods.
Microscopic counts
The most direct method of counting microorganism is by the use of a microscope and a slide with special chambers of known volume. These slides allow the counting of a small number of cells in a small volume and extrapolating the result to determine the population. An example of such a device is shown in Figure 30.3. A culture is placed on the slide marked with precise grids. The number of cells present in each grid is counted and an average determined. Conversion using a formula gives the number of cells per milliliter in the culture. This method is rapid, a result can be known in just a few minutes, and is easy to perform. However without the use of special viability stains, it is impossible to distinguish living cells from dead ones. If this distinction is important, direct microscopic counts are not the solution. Finally, cultures containing less than 1 million cells per ml are actually too dilute for direct counts since there are too few cells in the very small volume that is actually examined under the microscope for an accurate count.

Figure 30.3. A Petroff-Hausser counting chamber. By carefully counting the microbes in each square, it is possible to accurately determine the population of microbes in a sample.
Electronic particle counts - the Coulter counter
Electronic particle counters are useful if the number of bacteria in a sample needs to be counted on a routine basis. The method is based on the property that nonconductive particles, such as bacteria, cause a disruption in an electric field as they pass through it. A Coulter counter is a type of electronic particle counter in which there is a small opening between electrodes through which suspended particles pass, Figure 30.4. In this sensing zone, each particle displaces its own volume of electrolyte, causing a gap in the current. Such a current drop is recorded as one particle. By precisely controlling the rate at which solution passes through the opening, it is possible to get exact, reproducible counts at a rate of up to several thousand bacteria per second. Coulter counters are highly dependent upon particle size and are near their detectable limits with microorganisms. Particle counters that use light diffraction as a means of sizing and counting particles are also manufactured and can detect particle less than 1 µm in diameter.
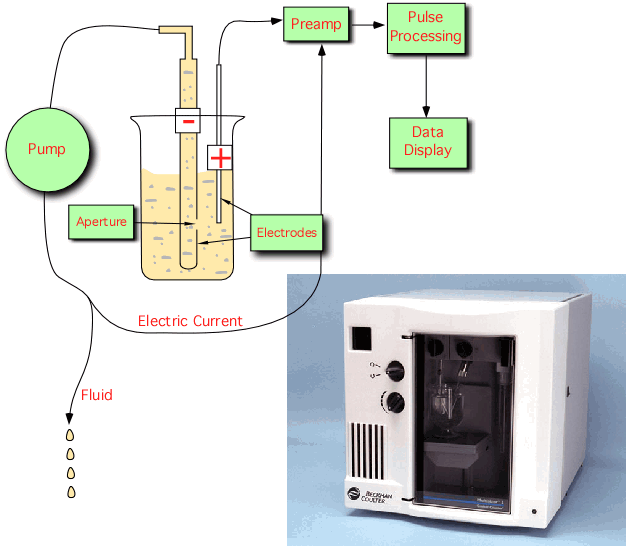
Figure 30.4. A coulter counter. Particle counters depend upon disruption of a current in a chamber. While there is an initial expense, they can save large amounts of time if many samples need to be counted. However, they cannot discriminate between microbes.
The advantage of this method is the simplicity of its operation and it reproducibility. As in microscopic counts, the machine cannot distinguish between living or dead cells or even between dust and bacteria. Any reasonably sized particle in the solution will be counted. There is also the expense of buying the counter, which can cost many thousands of dollars.
Viable counts
One of the most common methods of determining cell number is the viable plate count. Figure 30.5 shows a movie demonstrating the viable plate count. A sample to be counted is diluted in a solution that will not harm the microbe, yet does not support its growth (so they do not grow during the analysis). In most cases a volume of liquid (or a portion of solid) from the sample is first diluted 10-fold into buffer and mixed thoroughly. In most cases, a 0.1-1.0 ml portion of this first dilution is then diluted a further 10-fold, giving a total dilution of 100-fold. This process is repeated until a concentration that is estimated to be about 1000 cells per ml is reached. In the spread-plate technique some of the highest dilutions (lowest bacterial density) are then taken and spread with a sterile glass rod onto a solid medium that supports the growth of the microbe. It is important that the liquid spread onto the plate soaks into the agar. This prevents left over liquid on the surface from causing colonies to run together and the need for dry plates restricts the volume to 0.1 ml or less. A second method for counting viable bacteria is the pour plate technique, which consists of mixing a portion of the dilution with molten agar and pouring the mixture into a Petri plate. In either case are diluted so that individual cells are deposited on the agar and these give rise to colonies. By counting each colony, the total number of colony forming units (CFUs) on the plate is determined. By multiplying this count by the total dilution of the solution, it is possible to find the total number of CFUs in the original sample.
Figure 30.5. Viable Plate Count. Dilution plating and then spreading on an appropriate medium is a simple, yet effective way of determining the number of microbes in a sample. However, many microbes cannot grow in culture, so it does not give an accurate count of what is present in the environment.
One major disadvantage of the viable plate count is the assumption that each colony arises from one cell. In species where cells grow together in clusters, a gross underestimation of the true population results. One example of this is the genus Staphylococcus,, which is known to form clumps of microorganisms in solution. Each clump is therefore counted as one colony. This problem is why the term "CFUs per ml" is used instead of "bacteria per ml" for the results of such an analysis. It is a constant reminder that one colony does not equal one cell. Great care must also be taking during dilution and plating to avoid errors. Even one error in dilution can have large effects on the final numbers. The rate at which bacteria give rise to an observable colony can also vary. If too short an incubation time is used, some colonies may be missed. The temperature of incubation and medium conditions must also be optimized to achieve the largest colonies possible so that they are easily counted. Finally, this technique takes time. Depending on the organism, one day to several weeks might be necessary to determine the number of CFUs that were present when the experiment started. Such information may no longer be useful for many experiments.
Despite its shortcomings, the viable plate count is a popular method for determining cell number. The technique is sensitive and has the advantage of only counting living bacteria, which is often the important issue. Any concentration of microorganism can be easily counted, if the appropriate dilution is plated. It is even possible to concentrate a solution before counting, as is often done in water analysis, where bacterial populations w\are usually at low density. The equipment necessary for performing viable plate counts is readily available in any microbiology lab and is cheap in comparison to other methods. Finally, by using a selective medium it is possible to determine the number of bacteria of a certain class, even in mixed populations. These advantages have made viable plate counts a favorite of food, medical, aquatic and research laboratories for the routine determination of cell number.
Yet another method of determining the number of viable cells in a culture is called the most probable number method. By this method a culture is diluted to point where a certain small amount of that culture should contain approximately one cell, then multiple separate tubes of fresh culture medium are inoculated with aliquots of that small amount. After a suitable length of time, the tubes are checked to see how many of them display obvious bacterial growth. The fraction of tubes with growth then can be fitted to a curve to predict the actual number of viable cells in the starting culture, since the distribution of cells in the in the inoculated tubes must follow a Poisson distribution. Obviously, since we do not known exactly how much to dilute the culture before the samples are distributed into the tubes, several different dilutions must be tested. This way one of them will have a reasonable number of both "positive" and "negative" tubes. The precise nature of the curve fitting is more detailed than we need to review here, as is the statistical analysis that supports it.
Change in the amount of a cell component
In situations where determining the number of microorganisms is difficult or undesirable for other reasons, the use of indirect methods can be an excellent alternative. These methods measure some quantifiable cell property that increases as a direct result of microbial growth.
The simplest technique of this sort is to measure the weight of cells in a sample. Portions of a culture can be taken at particular intervals and centrifuged at high speed to sediment bacterial cells to the bottom of a vessel. The sedimented cells (called a cell pellet) are then washed to remove contaminating salt, and dried in an oven at 100-105 °C to remove all water, leaving only the mass of components that make up the population of cells. An increase in the dry weight of the cells correlates closely with cell growth. However, this method counts dead as well as living cells. There might also be conditions where the dry weight per cell changes over time or under different conditions. For example, some bacteria that excrete polysaccharides have a much higher dry weight per cell when growing on high sugar levels (when polysaccharides are produced) than on low. If the species under study forms large clumps of cells such as those that grow filamentously, dry weight is a better measurement of the cell population than is a viable plate count.
It is also possible to follow the change in the amount of a cellular component instead of the entire mass of the cell. This method may be chosen because determining dry weights is difficult or when the total weight of the cell is not giving an accurate picture of the number of individuals in a population. In this case, only one component of the cell is followed such as total protein or total DNA. This has some of the same advantages and disadvantages listed above for dry weight. Additionally, the measurement of a cellular component is more labor-intensive than previously mentioned methods since the component of interest has to be partially purified and then subjected to an analysis designed to measure the desired molecule. The assumption in choosing a single component such as DNA is that that component is relatively constant per cell. This assumption has a problem when growth rates are different because bacterial cells growing at high rates actually have more DNA per cell because of multiple initiations of replication.
Turbidity
A final widely used method for the determination of cell number is a turbidometric measurement or light scattering. Figure 30.6 shows an example of a spectrophotometer. This technique depends on the fact that as the number of cells in a solution increases, the solution becomes increasingly turbid (cloudy). The solution looks turbid because light passing through it is scattered by the microorganisms present and the turbidity is proportional to the number of microorganisms in the solution. The turbidity of a culture can be measured using a photometer or a spectrophotometer. The difference between these instruments is the type of light they pass through the sample. Photometers, such as the Klett-Summerson device, use a red, green or blue filter providing a broad spectrum of light. Spectrophotometers use prisms or diffraction gratings supplying a narrow band of wavelengths to the sample. Both instruments measure the amount of transmitted light, the light that makes it from the light source through the sample to the detector.

Figure 30.6. A Spec20D Spectrophotometer. A common, yet simple spectrophotometer made by Bausch and Lomb, the Spec 20D, is a reliable instrument for determining turbidity readings. After setting the wavelength and calibrating the machine, a sample is placed in the cuvette holder. The absorbance is read from the digital display.
When measuring light scattering it is important to consider the wavelength of light used a bacterial culture. Microorganisms may contain numerous macromolecules that absorb light, including DNA (254 µm), proteins (280 µm), cytochromes (400-500 µm), and possible cell pigments. When measuring bacteria by light scattering it is best to pick a wavelength where absorption is at a minimum and for most bacterial cultures wavelengths around 600 µm are a good choice. However, the exact wavelength chosen is species specific.
The amount of light transmitted through a sample is inversely proportional to cell number and can be expressed in the equation shown in Figure 30.7.
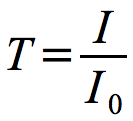
Figure 30.7. Transmittance. The relationship of transmittance to light entering and leaving the sample.
Where T is the light transmitted, I0 is the light entering the sample and I is the light passing through to the detector.
Due to the nature of light scattering, transmittance decreases geometrically as the cell numbers increase. It is more intuitive to think of the units increasing as growth increases and for most bacterial analysis, transmittance is converted into absorbance using the equation in Figure 30.8
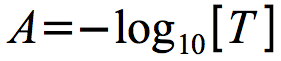
Figure 30.8. The definition of absorbance. Absorbance is defined as the negative log of transmittance.
Absorbance increases in a linear fashion as the cell number increases. When measuring growth of a culture the term optical density (OD) is normally used to more correctly represent the light scattering that is occurring; under optimal conditions, little light is actually absorbed by the culture so the term absorbance is misleading.
For most unicellular organisms changes in OD are proportional to changes in cell number (within certain limits) and therefore can be used as a method to follow cell growth. If a precise cell number for a given OD is desired, a standard curve can be generated, where viable plate count or cell mass is plotted as a function of OD. It also wise to develop a standard curve to verify that the OD is actually an accurate portrayal of cell growth. After the standard curve is made, it is then possible to simply measure the OD of the culture and read the cell number from the curve.
The turbidity of a culture is dependent upon the shape and internal light-absorbing components of the microorganism and therefore turbidity readings are species-specific and cannot be compared between different microbes or even between different strains of the same species. As above, there are microbes that change cell size or shape at different stages of growth, which introduces some inaccuracy to this method of cell counting. Also both living and dead cells scatter light and are therefore counted. However, the method is very rapid and simple to perform and provides reliable results when used with care, so it is an extremely common method of real time analysis of prokaryotic populations. Turbidometric measurements also do not destroy the sample.